BioProtIS: Facilitando a Análise de Interações entre Proteínas e Ligantes

A Revista Eletrônica PesquisABC possui o seguinte registro ISSN: 2675-1461
Graziela Sória Virgens¹,*, Júlia Oliveira¹, Maria Izadora Oliveira Cardoso¹, João Alfredo Teodoro¹, Danilo Trabuco do Amaral¹,*
1 Centro de Ciências Naturais e Humanas, Universidade Federal do ABC (UFABC), Santo André, São Paulo, Brasil.
*Endereço eletrônico: Este endereço de email está sendo protegido de spambots. Você precisa do JavaScript ativado para vê-lo.; Este endereço de email está sendo protegido de spambots. Você precisa do JavaScript ativado para vê-lo.
Resumo: A identificação de interações entre proteínas e ligantes é crucial para entender processos biológicos e descobrir potenciais bioprodutos. Para otimizar a descoberta de moléculas de interesse nas diferentes áreas das Ciências Biológicas, apresentamos o BioProtIS, um pipeline de Triagem Virtual Invertida (IVS) que integra análises de dinâmica molecular e docking. O objetivo principal é automatizar e agilizar a análise de interações proteína-ligante em escalas genômicas e transcriptômicas. Na era pós-genômica, a triagem computacional de alto rendimento tornou-se uma prática fundamental, oferecendo eficiência de custos e otimização do trabalho laboratorial. Contudo, há uma carência de pipelines que integrem eficientemente o processo de triagem virtual. O BioProtIS preenche essa lacuna, integrando ferramentas para facilitar o docking de ligantes com uma vasta gama de proteínas. Testamos o desempenho deste pipeline usando os transcriptomas de Cereus jamacaru (uma espécie de cacto) e Aspisoma lineatum (um vaga-lume), além do genoma de Homo sapiens. O BioProtIS permitiu, de maneira automatizada, a avaliação simultânea de múltiplos substratos com milhares de proteínas, oferecendo flexibilidade para customizações e a integração de novos algoritmos de docking. O acesso ao BioProtIS é gratuito e está disponível em : https://github.com/BBMDO/BioProtIS.
Palavras-chave: Docking; proteína-ligante; transcriptoma; Cereus; bioluminescente.
Abstract: The identification of interactions between proteins and ligands is crucial for understanding biological processes and discovering potential bioproducts. To optimize drug discovery, we present BioProtIS, an Inverted Virtual Screening (IVS) pipeline that integrates molecular dynamics analyses and docking. The main objective is to automate and expedite the analysis of protein-ligand interactions on genomic and transcriptomic scales. In the post-genomic era, high-throughput computational screening has become a fundamental practice, offering cost efficiency and optimization of laboratory work. However, there is a lack of pipelines that efficiently integrate the virtual screening process. BioProtIS fills this gap by integrating tools to facilitate the docking of ligands with a wide range of proteins. We tested the performance of the pipeline using the transcriptomes of Cereus jamacaru (a cactus species) and Aspisoma lineatum (a firefly), as well as the genome of *Homo sapiens*. BioProtIS improves accuracy in interactions and allows for the simultaneous evaluation of multiple substrates, offering flexibility for customizations and the integration of new docking algorithms. Access to BioProtIS is free and available at: https://github.com/BBMDO/BioProtIS.
Keywords: Docking; protein-ligand; transcriptome; Cereus; bioluminescente.
O que é o BioProtIS?
BioProtIS é uma sequência de programas alinhados que ajuda a analisar interações entre proteínas e substratos de maneira automatizada (Virgens et al., 2024)[1]. Em essência, essa ferramenta fornece um pipeline (processo automatizado que integra várias etapas), que facilita a análise e interpretação de dados relacionados a essas interações. Isso é especialmente importante em biologia molecular, biotecnologia e farmacologia, onde entender as conexões entre diferentes moléculas pode levar a descobertas significativas.
Passo a Passo do BioProtIS
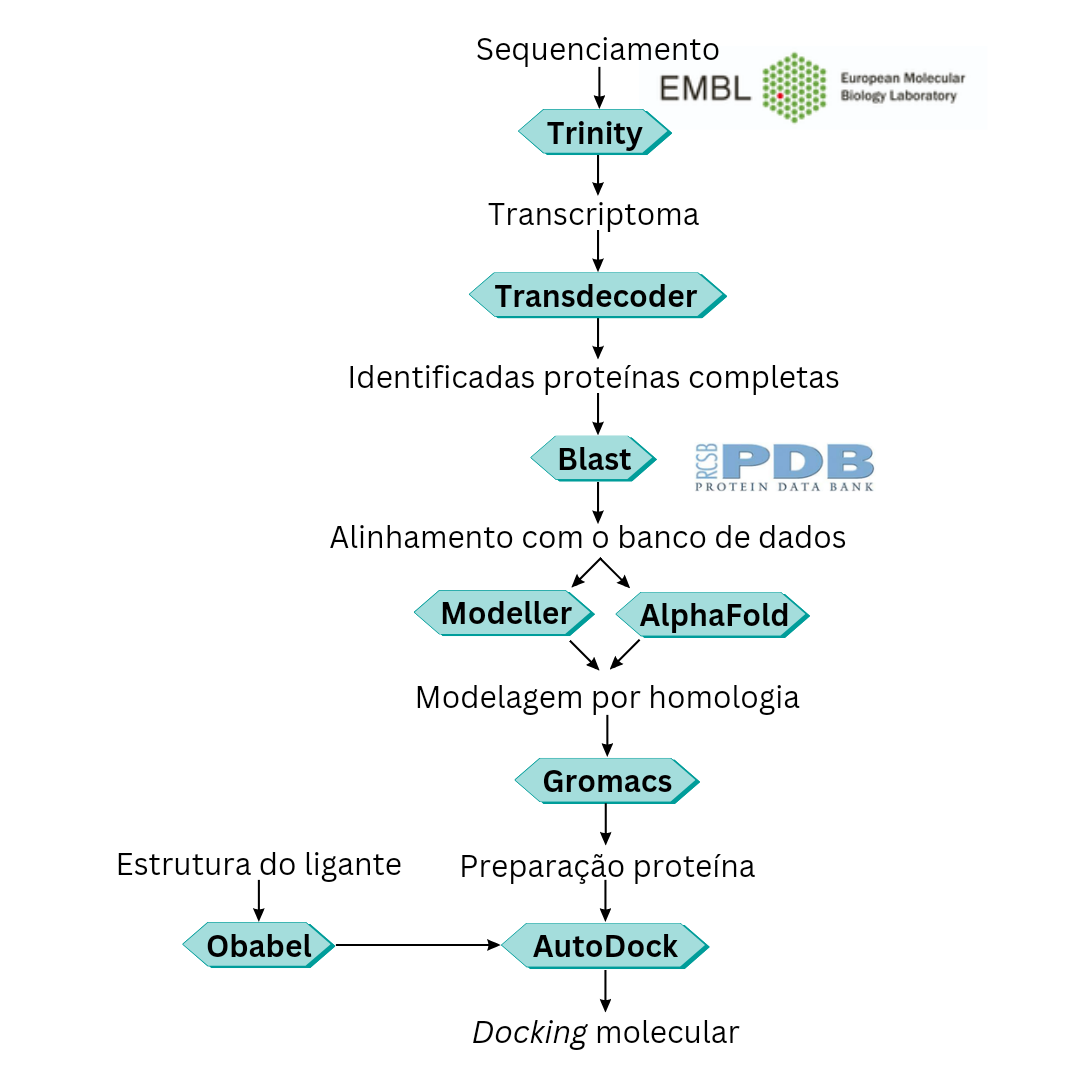
O processo de análise de interações proteína-ligante no BioProtIS envolve várias etapas (Figura 1), cada uma delas projetada para otimizar a eficiência e a precisão das análises. Vamos explorar cada passo do pipeline:
Passo 1: Identificação de Sequências Homólogas
No primeiro passo, utilizamos sequências de aminoácidos derivadas de montagens de transcriptoma (conjunto total de RNAs mensageiros expresso pelo organismo) e/ou genoma para realizar uma busca local por sequências homólogas. Essa busca é feita usando o programa BLAST, uma ferramenta que compara sequências de aminoácidos. As sequências são então organizadas em arquivos FASTA individuais para cada proteína identificada e agrupadas em pastas, permitindo uma estrutura organizada para a análise posterior.
Passo 2: Preparação de Dados para Modelagem de Proteínas
Após identificar as sequências homólogas, utilizamos um script fornecido pelo Protein Data Bank (PDB; banca de dados de estruturas 3D de proteínas) para baixar as estruturas de proteínas similares às sequências de aminoácidos estudadas. O pipeline está projetado para empregar, localmente, programas como Modeller (Webb et al., 2017)[2] ou AlphaFold2 (Jumper et al., 2020)[3] para materializar a proteína no formato 3D. Sequências com menos de 50% de identidade em relação ao modelo de referência são excluídas para garantir modelagem de maior qualidade.
Passo 3: Predição da Estrutura Tridimensional da Proteína
Aqui ocorre a modelagem 3D propriamente dita. Usamos Modeller ou AlphaFold2, dependendo dos recursos computacionais disponíveis. Geramos trinta modelos para cada proteína e selecionamos o modelo com a menor pontuação DOPE (Discrete Optimized Protein Structure) para etapas subsequentes. O modelo selecionado passa por refinamentos, incluindo otimização geométrica, minimização de energia e ajustes de resíduos, assegurando que a estrutura tridimensional gerada seja confiável e represente com maior precisão possível a proteína de interesse.
Passo 4: Análise de Docking Molecular
O que é Docking Molecular?
Docking molecular é uma técnica computacional utilizada para prever como uma molécula se liga a uma proteína alvo. O objetivo é identificar os locais de ligação e a afinidade entre a proteína e o ligante, o que é crucial para o desenvolvimento de novos medicamentos (De Vivo et al., 2016; Du et al., 2016)[4,5]
Após a obtenção dos modelos, usamos o Gromacs (Van Der Spoel et al., 2005)[6] para preparar as estruturas para a etapa de docking. Nesse passo, as estruturas de proteínas em formato PDB são processadas para criar um arquivo de topologia do Gromacs, essencial para estabelecer os parâmetros do ambiente em que ocorrem as interações da proteína durante as simulações. Para aliviar qualquer tensão estrutural, realizamos a minimização de energia. Essas etapas garantem que as estruturas estejam em condições adequadas para análises subsequentes.
Na análise de docking, o BioProtIS permite duas abordagens. Na primeira, utilizamos o software Fpocket (Le Guilloux et al., 2009)[7] para identificar os locais de ligação de substratos nas estruturas das proteínas (sítio de ligação ou sítio ativo. Essa metodologia sistematiza e permite a caracterização dos locais de ligação com base em propriedades como volume, forma, área de superfície e características hidrofóbicas/hidrofílicas. No segundo, o docking cego, permite que o programa de ancoragem busque a melhor configuração de pose e orientação do substrato junto a proteína, sendo interessante em estudos por busca de novos sítios ou sítios alternativos. Por fim, o docking de sítio ativo/ligação ou cedo são realizados pelo programa AutoDock Vina (Nguyen et al., 2019)[8], explorando os locais de ligação predefinidos ou alternativos.
Passo 5: Resumo dos Resultados
No último passo, selecionamos as proteínas com as menores energias de ligação aos substratos como potenciais alvos para estudos. Uma tabela é construída com os valores de energia e anotações funcionais, seguida pela criação de um histograma que avalia métricas globais.
Figura 1. Pipeline de etapas do BioProtIS.
Resultados das Aplicações
Os testes realizados com o BioProtIS demonstraram sua eficácia em prever interações proteína-ligante. Durante as validações, os pesquisadores utilizaram dados ômicos de uma espécie de cacto brasileiro, o Cereus jamacaru, de organismos bioluminescentes, e do próprio ser humano. As análises mostraram que as proteínas de interesse, envolvidas na bioluminescência, apresentaram uma clara afinidade por ligantes específicos.
Cereus
Para o Cereus, a plataforma permitiu identificar proteínas que participam na resposta a estresses ambientais e na produção de compostos bioativos. Os resultados indicaram que esses alvos possuem potenciais interações com compostos químicos presentes em ambientes naturais, o que pode ser útil para aplicações em biotecnologia.
No contexto cosmetológico, o projeto fez avanços na pesquisa de proteínas do C. jamacaru (mandacaru), focando nas interações dessas proteínas com substâncias como L-DOPA, que está relacionada ao surgimento de manchas na pele, e Acetil-CoA, que influencia a oleosidade. Usando esse sistema automatizado criado no laboratório, realizou-se simulações e modelos 3D para encontrar alvos promissores. Esse processo auxiliou na escolha das proteínas relevantes para entender melhor como elas interagem. Foram identificadas proteínas importantes, como peroxidases que estão envolvidas na regeneração da pele, e fosfatases serina/treonina que regulam várias funções celulares, incluindo o controle da produção de óleo na pele. Essas proteínas têm um grande potencial para aplicações na biotecnologia.
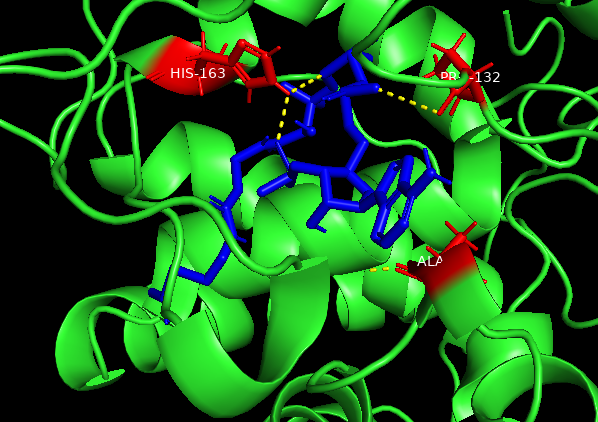
A figura 2 mostra uma representação tridimensional de uma dessas proteínas, a peroxidase ascorbato, responsável por reações antioxidantes nas células, representada pela cor verde. Em azul, está o substrato acetil-CoA, um composto importante para diversas reações celulares. Em vermelho (identificados como HIS-163 e HIS-112) são regiões da proteína que ajudam a manter o acetil-CoA no lugar certo para que a reação aconteça de forma eficaz (Ragsdale, 1991)[9].
Figura 2. Estrutura tridimensional da proteína 1apx - Cytosolic ascorbate peroxidase com o substrato Acetil-CoA.
Organismos bioluminescentes
No caso dos organismos bioluminescentes, o BioProtIS facilitou a identificação de estruturas proteicas envolvidas na síntese de luciferinas, conhecidas como luciferases, os compostos responsáveis pela luz nos seres vivos (Viviani, 2002)[10]. As análises de docking revelaram ligantes que poderiam se unir a essas proteínas com energias de ligação favoráveis, sugerindo uma base para o desenvolvimento de novos biossensores ou ferramentas diagnósticas baseadas em bioluminescência (Nobeli et al., 2009)[11].
Homo sapiens
Para o teste com o Homo sapiens, que possui mais de 20 mil genes (https://www.uniprot.org/proteomes/UP000005640), foi analisado proteínas que interagem com a ureia, que é produzida no fígado para eliminar a amônia, um subproduto tóxico do metabolismo, e transportada para os rins, onde é filtrada e excretada na urina (Matsumoto et al., 2019; Weiner et al., 2015)[12,13].
As proteínas com as dez melhores ligações com a ureia em termos de energia, mostraram estar envolvidas em regulação de expressão gênica, regulação da resposta imune, degradação de proteínas e sinalização celular, funções importantes para prevenir a toxicidade da amônia e manter o equilíbrio do corpo.
Os modelos gerados apresentaram baixas pontuações de DOPE, indicando que as estruturas tridimensionais eram estáveis e adequadas para estudos subsequentes. Os resultados do docking molecular revelaram potenciais ligantes que poderiam se ligar efetivamente às proteínas-alvo, com as menores energias de ligação indicando alta afinidade. Isso sugere que a ferramenta não apenas facilita a modelagem, mas também oferece insights valiosos para a pesquisa em farmacologia.
Por que as interações proteína-ligante são importantes?
As proteínas são cruciais em processos biológicos. Elas catalisam reações químicas, transmitem sinais e combatem infecções. Os ligantes, por sua vez, ligam-se a proteínas para desencadear ou inibir essas funções. Compreender essas interações é vital para o desenvolvimento de tratamentos para doenças, como câncer e diabetes (Cournia et al., 2017)[14].
O Impacto da Tecnologia na Pesquisa Biomédica e seu diferencial
Ferramentas como o BioProtIS, podem auxiliar os pesquisadores a acessarem e processar grandes volumes de dados de forma mais eficiente e sem a necessidade de conhecimento aprofundado em programação. Isso é relevante em um mundo onde a quantidade de informações cresce rapidamente. A capacidade de integrar dados de diferentes fontes e realizar análises complexas em um único ambiente pode levar a novas descobertas que, de outra forma, poderiam passar despercebidas.
Com o BioProtIS, o usuário pode utilizar as ferramentas de forma integrada para cada etapa, não necessitando de conversões a parte para ter uma continuidade no processo. Com este pipeline, as ferramentas estão em um único lugar gerando saídas e entradas para a próxima etapa de forma automatizada, exigindo apenas a inserção dos dados ômicos e dos substratos de interesse pelo usuário. Além do mais, este pipeline é totalmente adaptável a necessidade do usuário de acordo com onde ele quer chegar, caso não precise de todas as etapas.
Adaptabilidade do Sistema/Pipeline
O sistema foi desenvolvido com uma abordagem totalmente modular, permitindo sua adaptação às necessidades específicas de cada pesquisa ou projeto. Ele pode ser utilizado desde a etapa inicial, com apenas uma entrada de dados, até processos mais complexos, sendo aplicado em módulos ou etapas isoladas. Em nosso laboratório, esse pipeline tornou-se uma ferramenta central, integrando e facilitando diversas pesquisas, que evidenciam sua robustez e alta flexibilidade. Além disso, projetos de Iniciação Científica (IC) também têm se beneficiado significativamente dessa automação, gerando grandes volumes de dados e abrindo caminho para potenciais publicações em um futuro próximo. Esse pipeline não só otimiza o fluxo de trabalho, como também promove maior reprodutibilidade e eficiência nas análises, consolidando-se como uma ferramenta indispensável para nosso grupo de pesquisa.
Conclusão
Em suma, o BioProtIS representa um avanço significativo na análise de interações entre proteínas e ligantes, oferecendo uma ferramenta poderosa para pesquisadores em genômica e transcriptômica. Essa plataforma tem o potencial de acelerar a pesquisa e facilitar descobertas importantes, transformando o conhecimento científico em aplicações que beneficiem a sociedade. Com seus testes utilizando organismos como o Cereus e o vagalume, o BioProtIS não só demonstrou sua aplicabilidade em estudos básicos, mas também em aplicações práticas, abrindo caminhos para inovações que podem impactar diretamente a biotecnologia e a medicina. O futuro é promissor, e ferramentas como o BioProtIS estão na vanguarda dessa revolução na pesquisa biotecnológica.
Referências
1. Virgens, Graziela Sória, et al. "BioProtIS: Streamlining protein-ligand interaction pipeline for analysis in genomic and transcriptomic exploration." Journal of Molecular Graphics and Modelling 128 (2024): 108721.
2. Webb, Benjamin, and Andrej Sali. "Protein structure modeling with MODELLER." Functional genomics: Methods and protocols (2017): 39-54.
3. Jumper, John, et al. "AlphaFold 2." Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (2020).
4. De Vivo, Marco, et al. "Role of molecular dynamics and related methods in drug discovery." Journal of medicinal chemistry 59.9 (2016): 4035-4061.
5. Du, Xing, et al. "Insights into protein–ligand interactions: mechanisms, models, and methods." International journal of molecular sciences 17.2 (2016): 144.
6. Van Der Spoel, David, et al. "GROMACS: fast, flexible, and free." Journal of computational chemistry 26.16 (2005): 1701-1718.
7. Le Guilloux, Vincent, Peter Schmidtke, and Pierre Tuffery. "Fpocket: an open source platform for ligand pocket detection." BMC bioinformatics 10 (2009): 1-11.
8. Nguyen, Nguyen Thanh, et al. "Autodock vina adopts more accurate binding poses but autodock4 forms better binding affinity." Journal of Chemical Information and Modeling 60.1 (2019): 204-211.
9. Ragsdale, Stephen W. "Enzymology of the acetyl-CoA pathway of CO2 fixation." Critical reviews in biochemistry and molecular biology 26.3-4 (1991): 261-300.
10. Viviani, Vadim R. "The origin, diversity, and structure function relationships of insect luciferases." Cellular and Molecular Life Sciences CMLS 59 (2002): 1833-1850.
11. Nobeli, Irene, Angelo D. Favia, and Janet M. Thornton. "Protein promiscuity and its implications for biotechnology." Nature biotechnology 27.2 (2009): 157-167.
12. Matsumoto, Shirou, et al. "Urea cycle disorders—update." Journal of human genetics 64.9 (2019): 833-847.
13. Weiner, I. David, William E. Mitch, and Jeff M. Sands. "Urea and ammonia metabolism and the control of renal nitrogen excretion." Clinical Journal of the American Society of Nephrology 10.8 (2015): 1444-1458.
14. Cournia, Zoe, Bryce Allen, and Woody Sherman. "Relative binding free energy calculations in drug discovery: recent advances and practical considerations." Journal of chemical information and modeling 57.12 (2017): 2911-2937.
Redes Sociais